Bioaktive Peptidnaturstoffe in der Chemischen Biologie
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für molekulare Physiologie
Einleitung
Die vergleichsweise junge Disziplin der Chemischen Biologie widmet sich dem Studium und der Aufklärung biologischer Vorgänge mit chemischen Methoden. Besonders gesucht sind kleine Moleküle, deren Anwendung einen spezifischen biologischen Effekt hervorruft. Vielversprechend ist es dabei, sich Sekundärmetaboliten zuzuwenden, die von der Natur im Lauf der Evolution auf eine wichtige molekulare Funktion hin selektiert worden sind. Diese „Naturstoffe“ sind oft hoch bioaktiv und ermöglichen es, neue Wirkprinzipien aufzufinden und ihre Aktivitätsmuster auszunutzen. Von besonderem Interesse sind Peptidnaturstoffe (Abb. 1): Diese Klasse strukturell verwandter Substanzen überrascht mit einem sehr breiten Spektrum an unterschiedlichen Aktivitäten und umfasst Toxine wie Phalloidin (1), eines der Gifte des Knollenblätterpilzes, wichtige Antibiotika wie die Penicilline (2) und auch das Cyclosporin (3), dessen immunsuppressive Wirkung nach Organtransplantationen nahezu unverzichtbar ist. Über ihr enormes biologisches Potenzial hinaus fordern Peptidnaturstoffe auch die synthetische Chemie, die zur effektiven Darstellung und gezielten Modifikation solcher Moleküle weiterentwickelt werden muss. Das Studium dieser Substanzen an der Schnittstelle zwischen Chemie und Biologie kann daher beide Disziplinen befruchten.

Kartierung von Bindestellen durch chemisches Abtasten
Eine Aufgabe der Chemischen Biologie ist die Charakterisierung der Wechselwirkung von Peptidnaturstoff-Liganden mit größeren Biomakromolekül-Zielstrukturen, wie etwa Proteinen oder Nukleinsäuren. Neben der Bestimmung der Molekül-Affinitäten spielt die Klärung der räumlichen Lage und Orientierung des Liganden auf der Oberfläche der Makromoleküle eine wichtige Rolle. Zum Studium dieser Parameter unter möglichst realistischen Bedingungen kann die dem Liganden eigene chemische Reaktivität genutzt werden: Sie führt bei der Wechselwirkung der Moleküle mit einer Funktion der Zielstruktur zur Ausbildung einer kovalenten Bindung und zu einem analysierbaren „Schnappschuss“ der Orientierung. Bedingung ist, dass diese Reaktion nur bei großer räumlicher Nähe stattfindet (proximity induced covalent capture, PICC).
 In einem nichtkovalenten ternären Komplex aus L11-Protein, 23S-rRNA und Thiopeptid kann die Thiolgruppe (rot) von L11-Cysteinmutanten (hellgrau) mit einer Dehydroaminosäure (gelb) des Thiopeptids reagieren. Die Bildung eines kovalenten Addukts wird im Anschluss mittels Gelelektrophorese und Massenspektrometrie analysiert. B) Strukturen von Thiostrepton (4) und Nosiheptid (5) sowie die Reaktionen der Thiopeptid-Dehydroaminosäuren (grau) mit Protein-Thiolfunktionen (6/7). C) Visualisierung der biochemischen Abtastung an der Röntgenstrukturanalyse des ternären Komplexes von L11/23S-rRNA und Thiostrepton (links) bzw. Nosiheptid (rechts). Bereiche des L11-Proteins (hellgrün) mit reaktivem Potenzial sind rot gekennzeichnet, nicht-reaktive Positionen sind gelb markiert. Zur Orientierung wurden zwei für die Bindung wichtige RNA-Basen (A1067 und A1095) blau hervorgehoben.")
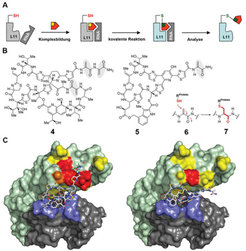
Eine solche Methode wurde zur Kartierung der Bindestelle der Thiopeptid-Antibiotika Thiostrepton (4) und Nosiheptid (5) entwickelt (Abb. 2), [1]. Dazu setzte man den Komplex aus ribosomaler RNA – 23S-rRNA – und dem ribosomalen Protein L11 ein, der die GTPase-Bindedomäne des bakteriellen Ribosoms bildet. Die Dehydroaminosäuren des Thiopeptid-Moleküls wurden mit künstlich hergestellten Cysteinmutanten des L11-Proteins zur Reaktion gebracht. Die Analyse ergab für diese Thiopeptide ein charakteristisch unterschiedliches Reaktivitätsmuster und ermöglichte so eine genaue Lokalisierung des Moleküls auf der Proteinoberfläche. Vergleiche mit kürzlich zugänglichen Röntgenstrukturanalysen der am Ribosom gebundenen Wirkstoffe zeigen eine hohe Zuverlässigkeit der Methode und lassen Schlüsse auf die Unterschiede zwischen dem Verhalten der isolierten biomolekularen Komponenten im Vergleich zum gesamten Ribosom zu.
Laufende Arbeiten konzentrieren sich nun auf die Ausweitung der Methode auf das intakte Ribosom sowie die Anwendung der Kartierungstechnik auf andere Kleinmolekül-Makromolekül-Interaktionen.
Thiopeptid-Naturstoffe als molekulare Sonden
Viele Wirkstoffe, die heute noch gegen Infektionskrankheiten eingesetzt werden, verlieren durch Resistenzen ihre Wirksamkeit. Darum müssen Wirkstoffe mit neuer Struktur und neuem Wirkprinzip gefunden werden. Eine Substanzklasse mit hohem antibakteriellen Potenzial sind die Thiopeptide, etwa Thiostrepton 4. Dieses Molekül bindet an das GTPase-assoziierte Zentrum des bakteriellen Ribosoms und unterbindet so gezielt die Proteinproduktion. Dieser Wirkmechanismus wird bislang in der Humanmedizin nicht genutzt.
 Struktur einer fluoreszenten Thiostrepton-Sonde (Fl: Fluorescein). B) Repräsentatives Diagramm der Quantifizierung des Bindungsverhaltens von Thiostrepton-Derivaten.")
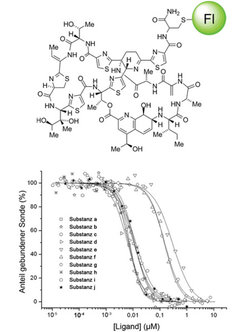
Durch chemische Synthese gelang es kürzlich [2], aus dem leicht verfügbaren Naturstoff Thiostrepton fluoreszente Sonden zu entwickeln, welche die eingehende Untersuchung des Bindeverhaltens von Antibiotika-Kandidaten am zellulären Wirkort des Thiostreptons erlauben (Abb. 3). So war es möglich, die Bindungseigenschaften verschiedener Substanzen quantitativ zu bestimmen. Vorteil des Verfahrens ist, dass die Sonden in automatisierten Testverfahren eingesetzt werden können und ihre Anwendung leicht auf Hochdurchsatzformate ausgeweitet werden kann. Dies wird die gezielte Entwicklung von neuartigen Antibiotika erleichtern, die den Wirkort des Thiostreptons ansteuern.
Im Rahmen der Forschungsarbeiten wurden darüber hinaus bereits einige Thiostrepton-Derivate synthetisiert, die hochspezifisch an den Wirkort des Naturstoffs binden. Manche dieser Substanzen zeigten viel versprechende Resultate in ersten Untersuchungen mit Pathogenen. Hauptgegenstand der laufenden Forschung ist nun die Synthese von Abkömmlingen der Thiostrepton-Struktur und anderer Thiopeptidderivate.
Synthesemethoden zur Darstellung von Thiopeptiden
Azoline

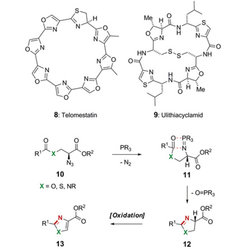
1,3-Azol(in)e und ihre Oligomere kommen häufig als wichtige strukturelle Elemente in Peptidnaturstoffen vor, wie etwa im Telomerase-Inhibitor Telomestatin (8) oder im Toxin Ulithiacyclamid (9). Durch Entwicklung einer Aza-Wittig-Methode für den Ringschluss zu 1,3-Azol(in)en (Abb. 4) gelang es den Dortmunder Wissenschaftlern [3], eine große Vielfalt an Oxazol(in)en (X = O) und Thiazol(in)en (X = S) zugänglich zu machen. Das milde und flexible Verfahren lässt reaktive funktionelle Gruppen in der Synthese zu und erlaubt zudem die Herstellung von heterozyklischen Dimeren. In laufenden Arbeiten konnte auch die Anwendbarkeit dieser Synthesestrategie auf die Herstellung von Imidiazol(in)en (X = NR) gezeigt werden. Die entwickelte Prozedur wird wegen ihrer Flexibilität die Herstellung komplizierter multiheterozyklischer Verbindungen mit hohem biologischen Potenzial ermöglichen. Die Anwendung auf bioaktive Zielmoleküle, insbesondere Thiopeptide wie Nosiheptid (5), wird aktuell bearbeitet.
3-Hydroxypyridine

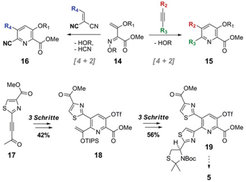
Um die komplexe Struktur von Nosiheptid 5 synthetisch zu erschließen, wurden zudem 1-Azadien-Hetero-Diels-Alder-Reaktionen entwickelt . Die Dortmunder Wissenschaftler konnten zeigen, dass sowohl Alkine [4] als auch Dicyano-Alkene [5] geeignete Substrate sind, 3-Hydroxypyridine regioselektiv zugänglich zu machen (Abb. 5). Quantenchemische Untersuchungen zum Reaktionsmechanismus lieferten das eindeutige Bild von konzertiert ablaufenden Zykloadditionen [5]. Durch Einsatz des Alkinylketons 17 konnte über den Enolether 18 der Synthesebaustein 19 erhalten werden, dessen weitere Umsetzung zu Nosiheptid 5 zurzeit studiert wird.
Synthese und Evaluierung von Chondramid C
Nicht nur das antibiotische, sondern auch andere Wirkprinzipien sind von Interesse. Der Peptidnaturstoff Chondramid C (20) etwa stabilisiert die polymere Form des Zytoskelettproteins Aktin (das F-Aktin) und greift so in die Reorganisationsprozesse der Zellstruktur ein. Im Gegensatz zum Pilzgift Phalloidin 1, das einen ähnlichen Wirkmechanismus aufweist, durchdringt Chondramid C die Zellmembran wesentlich leichter und eignet sich daher sowohl als chemisch-biologisches Werkzeugmolekül, als auch als Leitstruktur für die Entwicklung von auf Aktin gerichteten Wirkstoffen.
 Totalsynthese und Strukturbeweis von Chondramid C durch festphasengestützten Aufbau und E-selektive Ringschlussmetathese. B) Fluoreszenzmikroskopische Abbildung (Balkenlänge: 10 μm) von nicht mit Wirkstoff behandelten BSC-1-Zellen (Affenniere), rot: F-Aktin, blau: DNA. C) Abbildung von BSC-1-Zellen im gleichen Maßstab nach Behandlung mit Chondramid C (200 nM). Die Ausbildung eines durch Aggregation von Aktinfasern (tiefrote Bereiche) hervorgerufenen, viel kompakteren Phänotyps ist deutlich zu erkennen. D) Durch \"Docking\" erhaltenes molekulares Strukturmodell (Balken: 0.1 nm) der Bindung von Chondramid C (grün) an F-Aktin (grau) im Vergleich mit dem Pilzgift Phalloidin (gelb).")
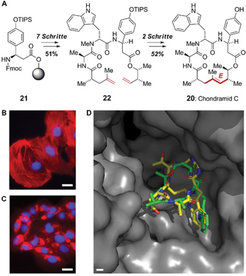
Zur Untersuchung des aus natürlichen Quellen nicht in größeren Mengen zugänglichen Chondramids wurde zunächst eine effiziente festphasengestützte Synthese entwickelt, in der am Ende der Synthesesequenz durch eine Ringschlussmetathese der entscheidende Ringschluss gelang – direkt unter Aufbau der gewünschten E-konfigurierten Doppelbindung (Abb. 6). In gleicher Weise erhielten die Forscher auch verschiedene Derivate, die sie auf ihre Wirkung in Zellen hin charakterisierten. Durch computergestütztes "Docking" konnte darüber hinaus ein Vorschlag für die Bindegeometrie von Chondramid C an F-Aktin abgeleitet werden. Es wird erwartet, dass aus diesen Daten eine neue Generation zellgängiger und potenter Aktinmodulatoren erwachsen kann [6].
Ausblick
Das gezielte Studium von Peptidnaturstoffen liefert in verschiedenen Gebieten der Chemischen Biologie wesentliche Beiträge. Die inhärente strukturelle Ähnlichkeit dieser Verbindungen wird chemische Biologen in Zukunft sicher dazu anregen, vollkommen neuartige Molekülstrukturen anzustreben und im Hinblick auf spezifische biologische Fragestellungen maßzuschneidern. Die so erhältlichen Moleküle werden das Verständnis über die molekularen Prozesse in der Zelle verbessern und können in Zukunft auch in Testverfahren und neuartigen Therapieansätzen eine Rolle spielen – gerade auf dem Feld der Infektionskrankheiten.