Computerunterstützte Experimente lassen Zellen leuchten, um uns zu erleuchten
Forschungsbericht (importiert) 2012 - Max-Planck-Institut für molekulare Physiologie
Einleitung
Ob Einzeller oder Teil eines komplexen Organismus – um lebensfähig zu sein, muss eine Zelle in der Lage sein, mit ihrem Umfeld zu interagieren. Sie muss Botenstoffe registrieren und interpretieren, auf Veränderungen ihrer Umgebung reagieren können und mit den zur Verfügung stehenden Ressourcen haushalten. Hierzu stehen jeder Zelle Proteine zur Verfügung, die diese vielfältigen Aufgaben über komplizierte Wechselwirkungen untereinander bewältigen können. In den letzten Jahren hat sich immer mehr herausgestellt, dass es nicht ausreicht, die statische Struktur eines durch ein Gen kodierten Proteins zu verstehen, da die in Zellen produzierten Proteine im ständigen Wandel sind. So ist unter anderem ihr Aufenthaltsort in der Zelle von entscheidender Bedeutung, denn gerade die Möglichkeit, die räumliche Trennung zweier Bindungspartner zu regulieren, macht ihre Interaktion erst kontrollierbar. Dieses Phänomen wird zum Beispiel in Signalwegen ausgenutzt, welche – ausgelöst durch Botenstoffe – den Befehl zur Teilung, aber auch zum „Selbstmord“ (Apoptose) einer Zelle erteilen können. So kann eine Mutation des Gens, welches das an einem Signalweg beteiligte Protein kodiert, gesunde Zellen in Tumorzellen verwandeln.
Im Zentrum der Aufmerksamkeit einer Gruppe am MPI für molekulare Physiologie steht das berüchtigte Produkt eines Krebsvorläufergens, das sog. Ras-Protein (von: Rat Sarcoma). In mutierter Form ist es in etwa 30 Prozent aller Krebsarten zu finden, in Bauchspeicheldrüsenkrebs sogar in 90 Prozent aller Tumore, und ein Vorhandensein seiner mutierten Form verhindert zum Beispiel bei Darmkrebs eine ansonsten erfolgversprechende EGFR-Antikörper-Therapie [1]. Innerhalb einer Zelle wird Ras an der Zellmembran angereichert, was seine zentrale Rolle in verschiedenen Signalwegen begründet. Allerdings befindet es sich in ständigem Austausch mit anderen Bereichen und Organellen der Zelle. Tatsächlich müsste sich Ras eigentlich unspezifisch über die gesamte Zelle verteilen. Die Zelle muss also für die unsymmetrische Lokalisierung von Ras an der Zellmembran einen Mechanismus bereitstellen und Energie aufwenden. Diese Lokalisierungsstruktur muss aktiv aufrechterhalten werden und stellt eine Art von selbstorganisierter Musterbildung im Größenmaßstab der Zelle dar. Sie entsteht allerdings durch die molekularen Wechselwirkungen zwischen den Proteinen und kann nicht verstanden werden, wenn man nur die Interaktionspartner von Ras kennt. Die Wechselwirkungen zwischen Proteinen entwickeln eine Dynamik, die für diese Selbstorganisation verantwortlich ist. Nur die Erforschung dieser dynamischen Prozesse ermöglicht es zu verstehen, warum eine Zelle Energie aufwendet, um ein Muster zu kreieren, und wie auf diese Musterbildung Einfluss genommen werden kann. So gibt es Krebszellen mit mutiertem Ras, deren Überleben von der Ras-Lokalisierung an der Zellmembran abhängt. Gelingt es, diese Lokalisierung gezielt zu stören, so kann man diese Zellen in den Selbstmord treiben.
Die Zelle kann mit Proteinen rechnen
Die an den diversen Interaktionen in der Zelle beteiligten Proteine ändern sich ständig. So werden kleine Moleküle angebunden, Teile der Proteine abgespalten oder permanent mit anderen verbunden, oder es bilden sich Komplexe aus mehreren Proteinen. Der kollektive Zustand der Proteine definiert den Zustand der Zelle, zum Beispiel als Reaktion auf die extrazelluläre Umgebung. Der Zustand der Proteine, das heißt welche Proteine gerade aktiv oder überhaupt vorhanden sind, und die Dynamik ihrer Wechselwirkungen untereinander sind derart eng miteinander verwoben und nehmen bidirektional aufeinander Einfluss, dass Konzepte von Ursache und Wirkung nicht mehr greifen. Diese in sich geschlossene Rückkopplung erlaubt es der Zelle, aus dynamischen, lokalen Prozessen globale Muster zu generieren, wie es schon Alan Turing für gekoppelte chemische Reaktionen vorhergesagt hat [2]. Aus dem Verhalten der Proteine errechnet eine Zelle Muster, welche ihr „Gedächtnis“ repräsentieren und ihre Funktion definieren. Auf diese Art können kurzlebige Reize von außen in langfristige Änderungen der Form und des Verhaltens einer Zelle übersetzt werden.
Auf vergleichbare Art versucht die Systemische Zellbiologie, das Detailwissen aus verschiedenen wissenschaftlichen Sparten zu einem zusammenhängenden Bild der Zelle und ihrer Funktion zu formieren. Zum Beispiel erlaubt eine detaillierte Analyse des Erbguts, welches für Proteine kodiert, abzuschätzen, welche Proteine überhaupt miteinander interagieren können. Allerdings entzieht sich das Spektrum der möglichen Modifikationen, die ein Protein tatsächlich durchläuft, der Vorhersage und muss im zellulären Kontext experimentell bestimmt werden. Ebenso reicht dieses Wissen aller möglichen Modifikationen alleine nicht aus, um zu erklären, auf welche Art die Modifikation eines Proteins seine Wechselwirkung mit anderen beeinflusst und wie dies wiederum die Musterbildung innerhalb der Zelle reguliert. Diese einzelnen, aber ineinandergreifenden Bausteine müssen erst verknüpft werden, um die Zelle als einheitliches System zu verstehen.
Wie eine Zelle auf Signale reagiert
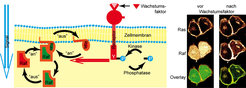
Für die essenzielle Aufgabe der gesteuerten Zellteilung bildet der Organismus gezielt Botenstoffe, sogenannte Wachstumsfaktoren, wie zum Beispiel den Epidermal Growth Factor EGF. Wird ein Wachstumsfaktor von einem für ihn vorgesehenen Rezeptor, der in der Zellmembran sitzt, gebunden, so verändert dies den Rezeptor. Das wiederum ermöglicht anderen Proteinen, den Rezeptor als Bindungspartner zu erkennen und ihn – im Falle von sogenannten Kinasen – durch Phosphorylierung (das Anhängen einer Phosphatgruppe) weiter zu verändern. Um die Information von den aktiven Rezeptoren in die Zelle zu tragen, wird das kleine G-Protein Ras „aktiviert“, welches in der Zellmembran mobiler ist als der Rezeptor. In seiner aktiven Form wird es von weiteren Proteinen (z.B. Rat Fibrosarcoma Raf) erkannt. Wie beim Dominospiel entsteht eine Kette aus Modifikationen verschiedener Proteine, welche die Information ins Innere der Zelle weitertragen [3] (Abb. 1). Die Maschinerie der Signalweiterleitung gleicht allerdings einem laufenden Motor, der nur darauf wartet, dass ein Gang eingelegt wird, denn die beteiligten Proteine und ihr Zustand befinden sich in ständigem Fluss. Wie beim Gasgeben und Bremsen gibt es für jede Veränderung im Zustand der Proteine einen Gegenspieler. So gibt es z. B. für die phosphorylierten Rezeptoren sogenannte Phosphatasen, welche die Phosphorylierung rückgängig machen. Ob ein Rezeptor ruht oder aktiv ist, ergibt sich dann durch die Verschiebung der Balance zwischen der Phosphorylierung und der Dephosphorylierung, vergleichbar dem Yin und Yang in der Körperbalance. Um diese Balance geregelt zu manipulieren, sind viele Wege denkbar, beispielsweise ein Herunterregeln der Phosphataseaktivität, bevor sie mit den Rezeptoren interagieren kann, oder ein Verstärken der Kinaseaktivität durch Erhöhen ihrer Konzentration in der Nähe der Rezeptoren.
: Binden eines Wachstumsfaktors „aktiviert“ den Rezeptor, der Ras anschaltet, sodass Raf gebunden werden kann, was wiederum Raf aktiviert und so das Wachstumsfaktorsignal tiefer in die Zelle übermittelt. Jeder der Pfeile stellt eine durch Proteine geregelte Reaktion dar. Lebende Zellen unter dem Fluoreszenz-Mikroskop (rechts): Die Proteine Ras und Raf wurden mit zwei unterschiedlichen Fluorophoren markiert und eine Aufnahme der Zelle vor und nach Stimulation mit dem Wachstumsfaktor EGF gemacht. Man erkennt deutlich, dass Ras an der Plasmamembran, aber auch an einer Struktur innerhalb der Zelle (dem Golgi-Apparat) angereichert ist. Nach Stimulation wird Raf von aktiviertem Ras rekrutiert und dupliziert so das Muster, das Ras gebildet hat. Wie man an der Rekrutierung von Raf an den Golgi-Apparat erkennen kann, ist aktives Ras auch im Inneren der Zelle zu finden, obwohl es dort weder EGF noch EGF-Rezeptoren gibt. Auf diese Art nutzt die Zelle den dynamischen Lokalisierungsmechanismus von Ras, um das Aktivierungssignal von EGF im Inneren der Zelle zu verbreiten.")
Die Zelle unter dem Mikroskop
Um Einblick in den Mikrokosmos der molekularen Dynamik zu erhalten, wurden Messmethoden konzipiert, die Proteinkonzentrationen oder auch Protein-Wechselwirkungen in lebenden Zellen sichtbar machen. Die Fluoreszenzmikroskopie erlaubt es unter anderem, sowohl einzelne Moleküle bei der Diffusion zu beobachten, als auch den Aufenthaltsort und die Wechselwirkung zwischen nahezu beliebigen Paaren von Proteinen nachzuvollziehen. Hierbei werden spezielle Proteine (Fluorophore), die bei Bestrahlung mit Licht bestimmter Wellenlänge zur Emission von Licht mit anderer Wellenlänge angeregt werden, an interessante Proteine gekoppelt und können dann unter dem Mikroskop verfolgt werden. Eine wichtige Methode, um Informationen über das Verhalten der Proteine zu erhalten, besteht darin, eine Störung (Perturbation) des aktuellen Leuchtzustandes zu erzeugen, und nachzuvollziehen, wie sich ein neues Fließgleichgewicht einstellt. So gibt es zum Beispiel Proteine wie Ras, die fast ausschließlich in der Zellmembran zu sehen sind. Schaltet man die Lichtemission eines an ein solches Protein gekoppelten Fluorophors selektiv in nur einem Teil der Zellmembran an, kann man verfolgen, wie sich die Fluoreszenz über den Rest der Membran verteilt, und ebenso, ob diese nun leuchtenden Proteine noch in anderen Organellen der Zelle auftauchen.
Datenauswertung durch Simulation: Auf zu neuen Experimenten
Möchte man nun Messungen des molekularen Lebens verschiedener Zellen miteinander vergleichen, so muss man etliche Faktoren beachten: wie groß sind die Zellen, an welcher Stelle relativ zur Form der gesamten Zelle wurde die Perturbation durchgeführt, wieviel Zellmembran hat jede Zelle im Verhältnis zu ihrem Volumen, welche Konzentration des Fluorophors hat die Zelle produziert, und viele mehr. Einfache mathematische Modelle sind unzureichend, um solche Perturbationsexperimente vollständig zu beschreiben. Daher ist eine sinnvolle Auswertung nur möglich, wenn die Geometrie der Zelle gleichzeitig mit der Perturbation erfasst und die Zelle dann im Computer virtuell nachgebildet wird. Diese Computersimulation einer Zelle wird mit virtuellen Proteinen gefüllt, deren Eigenschaften durch Simulationsparameter gesteuert wird (Abb. 2). Wiederholt man nun Hunderte von Simulationen mit gezielt variierenden Parametern und vergleicht jedes Mal Simulation und Experiment, so erhält man schließlich einen Satz biologisch relevanter Parameter, wie Diffusionskonstanten oder Assoziations- und Dissoziationsraten, der das Experiment so gut wie möglich beschreibt. Um diese rechenaufwändige Aufgabe für viele Experimente durchführen zu können, bieten sich sogenannte „Zelluläre Automaten“ an. Diese Simulationstechnik wurde schon auf makroskopische biologische Systeme angewandt [4], aber hier wird sie zum ersten Mal auf intrazellulärer Ebene eingesetzt. Auf dieser Skala werden Interaktionen von Proteinen durch „Verhaltensregeln“ beschrieben. Für jedes Experiment liegt eine virtuelle Zelle vor, generiert aus tatsächlichen Messdaten biologischer Experimente. Dies erleichtert die Durchführung von Gedankenexperimenten durch Simulation enorm: Was wäre, wenn Protein A mit Protein B nur an der Zellmembran interagiert oder Protein B durch Protein C in seiner Funktion gestört wird? Diese Möglichkeit, Hypothesen in einem realistischen Umfeld zu testen, stellt eine der Stärken dieses Modellierungsansatzes dar. Hierdurch wird es möglich, neue Experimente zu konzipieren, welche die in der Simulation gewonnene Einsicht in das Netzwerk von Protein-Wechselwirkungen integrieren. [5]
. Ein gezieltes Anpassen dieser Werte in der Simulation liefert nach vielen Iterationen eine Übereinstimmung mit den experimentellen Messdaten und somit die biologisch relevanten Parameter.")
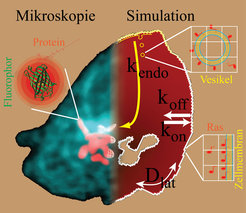
Ras im Mittelpunkt: ein Protein auf der Strafbank und im Mittelfeld
In dem obigen Beispiel eines aktivierten Rezeptors spielt ein Mitglied der Familie der „kleinen G-Proteine“, das Ras, eine zentrale Rolle. Es ist im Ruhezustand „aus“ und kann von aktivierten Rezeptoren über weitere Hilfsproteine „an“-geschaltet werden. Im angeschalteten Zustand wird es zum Beispiel von der Kinase Raf als Bindungspartner erkannt und leitet das Rezeptor-Aktivierungssignal weiter, indem es Raf an die Zellmembran rekrutiert. Rezeptoren verfügen meist über eine Signalsequenz, die Transportproteine veranlasst, sie in die Zellmembran einzubetten. Ras fehlt eine solche direkte Integrationsmöglichkeit, es befindet sich dennoch stark angereichert in der Zellmembran. Dies erleichtert seine Funktion, da es für aktivierte Rezeptoren einfach zu finden sein sollte, aber der genaue Mechanismus für die energieaufwändige Anreicherung an der Zellmembran wurde von uns erst kürzlich für HRas erklärt [6] und soll nun für das verwandte KRas verstanden werden, dessen mutierte Form bei vielen Krebsarten zu finden ist. Auch bei diesem dynamischen System ist wieder das Yin und Yang zwischen entgegengesetzten Aktionen ausschlaggebend. Ras wechselt zwischen einer trägen Form, der es schwer fällt, die Zellmembran zu verlassen (Trapping auf der Strafbank), und einer sehr mobilen Form (Exploration im Mittelfeld), der es möglich ist, die Zelle schnell zu erkunden, falls sich das Ras-Molekül „verirrt“ hat. Nur so kann Ras an der Zellmembran angereichert werden. Die „Verirrung“ von Ras wird dadurch begünstigt, dass die Zellmembran selber keine statische Struktur darstellt, sondern mit dem Zellinneren ständig Vesikel austauscht, auf denen auch Ras sitzt. Malte Schmick und sein Team untersuchen mit der oben beschriebenen Simulationstechnik, welche Proteine diesen Wechsel zwischen Trapping und Exploration begünstigen und welche Eigenschaften von Ras selbst hierfür wichtig sind.
Da das Netzwerk zur Ras-Lokalisierung offenbar unabhängig von dem Netzwerk zur Ras-Aktivierung und Signalweiterleitung ist, bieten sich hier neue Angriffsmöglichkeiten zur Krebsbehandlung. Denn Ras existiert in vielen Krebsarten in einer mutierten, permanent angeschalteten Form. Dies wiederum schließt verschiedene Signalwege kurz und die betroffene Zelle verliert die Fähigkeit, auf Botenstoffe korrekt zu reagieren. Die neuesten Ergebnisse zeigen die Möglichkeit auf, Krebszellen, die von einer Anreicherung des mutierten KRas an der Zellmembran abhängig geworden sind, in die Apoptose zu treiben. Hierzu reicht es aus, in den Lokalisierungsmechanismus von Ras einzugreifen, ohne auf die Signalwege direkt Einfluss nehmen zu müssen.