
Stefan Raunser
Direktor, Strukturbiochemie
Research Concept
Cellular processes result from the sophisticated but controlled interaction of many proteins that often assemble into large macromolecular complexes to optimally develop their function. The knowledge of the underlying mechanisms at molecular detail is important to get deep insight into the complex dynamics of the cell and to understand its disturbance in disease. We use stain and cryo-electron microscopy (cryo-EM) and biochemical techniques to study protein structures and to obtain their function.
The Structural Basis of Muscle Contraction

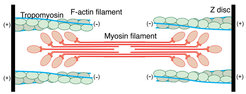
For almost 9 years, our group has been interested in filamentous actin (F-actin) and its function in muscle fibers and the eukaryotic cytoskeleton [1-6]. Our ultimate aim is to understand how F-actin interacts with a plethora of proteins to perform different essential cellular functions. In particular, we want to elucidate the structural basis of muscle contraction and its regulation. In addition, we want to decipher the architecture of the complete sarcomere. Sarcomeres, which are the repeating units of striated muscle fibers, are composed of thick myosin and thin actin filaments that slide along each other during muscle contraction (Figure 1).
 decorated with tropomyosin (yellow). Middle: Close-up view of a with the atomic and molecular model of an F-actin subunit (cyan) and tropomyosin (yellow) and their corresponding densities, respectively. The density corresponding to ADP is depicted in red. Right: Structure of the ATM complex. Cryo-EM reconstruction of F-actin (five central subunits in green and cyan) decorated with tropomyosin (blue) and myosin (central molecules in red).")
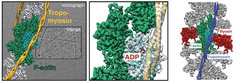
Since actin filaments resisted crystallization so far, fiber diffraction and cryo-electron microscopy have been the only methods to obtain structural information of F-actin and its interacting proteins. Using a combination of modern techniques, own optimized reconstruction programs [7], optimal samples as well as state-of-the-art equipment, we previously obtained a 3D reconstruction of the actin-myosin-tropomyosin complex at an unprecedented resolution of 8 Å [5]. Building on our established methods and applying the technology of direct electron detectors and cryo-EM, we now for the first time obtained high-resolution structures of F-actin in complex with tropomyosin (3.7 Å) (von der Ecken et al. 2015) and of F-actin in complex with tropomyosin and rigor state myosin using cryo-EM (3.9 Å) (von der Ecken et al. 2016) ( Figure 2). Noteworthy, this is more than 25 years after the crystal structure of G-actin was solved [8].
The structures revealed important details of intra- and interstrand interactions stabilizing the F-actin filament. The D-loop of F-actin is ordered and acts as a key mediator during filament formation. The actomyosin structure revealed details of the actomyosin interface, which is mainly stabilized by hydrophobic interactions. Surprisingly, the overall structure of myosin is similar to rigor-like myosin structures in the absence of F-actin, indicating that F-actin binding induces only minimal conformational changes in myosin.
Our results serve as a strong foundation for the molecular understanding of many cytoskeletal diseases, such as autosomal dominant hearing loss and diseases affecting skeletal and cardiac muscles, in particular nemaline myopathy and hypertrophic cardio-myopathy.
Bacterial Toxin Complexes

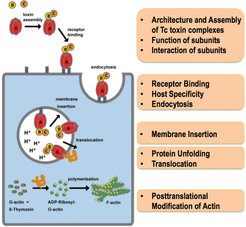
Another major interest of our department is to understand the function and structural organization of bacterial toxin complexes that permeate the membrane and translocate actin-attacking enzymes into the target cell. Bacterial ABC-type toxins are important virulence factors of a range of bacteria, including Photorhabdus luminescens [9] and Yersinia pseudotuberculosis [10] that infect insects and humans. Belonging to the class of pore-forming toxins, tripartite Tc complexes perforate the host membrane by forming channels that translocate toxic enzymes into the host (Figure 3). Our overall goal is to completely understand the mechanism of Tc toxin action (Figure 3).
In our initial cryo-EM work on the P. luminescens Tc complex we discovered that Tcs use a special syringe-like device for cell entry (Gatsogiannis et al. 2013) ( Figure 4). Building on these results, we unraveled the molecular mechanism through which this unusual and complicated injection system allows membrane permeation and protein translocation. We extended our research to X-ray crystallography complemented by cryo-EM to obtain structures of the 1.7 MDa toxin at atomic resolution. We obtained well-diffracting crystals of the TcA as well as of the TcB and TcC subunits and solved the structures by molecular replacement using our cryo-EM structure as model (Meusch et al. 2014).
The structures revealed that TcB and TcC (TcdB2 and TccC3) form a cocoon, in which the C-terminal region of TcC is autoproteolytically cleaved. Pentameric TcA forms a translocation channel, which is surrounded by a shell that comprises four putative receptor-binding sites and a neuraminidase-like region, probably important for the host specificity of the respective toxins. In addition, we showed that a pH-induced opening of the shell releases a putative entropic spring that drives the injection of the TcA channel into the membrane. Binding of TcB/TcC to TcA opens a gate formed by a six-bladed ß-propeller and results in a continuous protein translocation channel. This mechanism is unique and specific for Tc toxins and differs from that of other PFTs.


In our most recent work (Gatsogiannis et al. 2016) we have embedded TcA in nanodiscs as we have pioneered with the RyR1 receptor (Efremov et al. 2015) to ensure a natural lipid environment and stability of TcA. We then used cryo-EM with direct electron detection and single particle analysis to determine the structure of TcA in its pore state. The 3.5 Å structure reveals that the transmembrane helices rearrange in the membrane and open the initially closed pore, similarly to an iris diaphragm. However, they do not span the membrane completely. The edge of the channel is formed by extended loops instead that interact with lipid head groups and reach just to the surface of the membrane. To our knowledge, it is the first time that the transmembrane region of a Tc toxin and in general of a PFT is described in its near-to-native environment in atomic detail.
In addition to Tc toxins, we also work on other toxin complexes. For instance, our structural investigation of the Type VI secretion system in collaboration with J. Mougous, HHMI, University of Washington, provided important mechanistic insight into intercellular membrane protein delivery between bacteria (Whitney et al. 2015).
Functional and Structural Investigation of Membrane Proteins

Membrane proteins are key pharmacological targets and essential for many cellular functions. Since my master studies at the University of Mainz in 1999, I have been interested in the functional and structural investigation of membrane proteins [11-13]. When we started our group at the MPI in Dortmund in 2008, we set out to elucidate the structure-function relationship of different enzymes involved in the regulation of cellular processes.
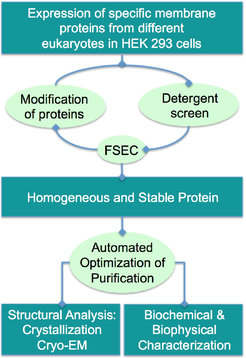
We use a mammalian expression system to express the proteins and established a complete high-throughput workflow to purify the proteins (Figure 5). As we have done for the ryanodine receptor RyR1, the structure of which we have solved in two different states using cryo-EM (Efremov et al. 2015) ( Figure 6), we plan to examine these proteins in lipid nanodiscs to provide a near-to-native membrane environment. At the moment, many membrane proteins are under investigation in the lab and prove to be useful for cryo-EM. Besides structural studies, we are establishing functional assays to study and characterize these proteins in vivo and in vitro.
 to the EF-hand domain (purple) induces conformational changes that are transmitted to the domains underneath the α-solenoid 1 (orange) and the C-terminal domain (red).")

In addition to our main projects we are involved in several side projects (mainly in collaboration with other groups), studying the molecular architecture of tethering complexes [14-16], septin complexes [17, 18], vesicle fusion [19], kinetochore complexes [20-23] and hemocyanin complexes (Gatsogiannis et al. 2015).
SPHIRE – Solving High Resolution Cryo-EM Structures

In collaboration with P. Penczek at UT Houston, we completely reworked the powerful cryo-EM image processing package SPARX [24]. The new program, called SPHIRE for Sparx for High Resolution Electron Microscopy, is open source and online since fall 2016 (sphire.mpg.de). Sphire is streamlined, easy to use, and ideally suited for solving high-resolution cryo-EM structures through minimal user interaction via a graphical user interface (Figure 7). We devised various statistical evaluation methods for the quality-assessment and validity of 3D reconstruction. Specifically, the concept of reproducibility by statistical resampling is incorporated into major processes of single particle analysis. Importantly, the program is not a black box and experienced users still have the possibility to perform their individual fine-tuning, which is especially useful for non-standard protein complexes.
Literature
1. Gao, M., Berghaus, M., von der Ecken, J., Raunser, S., and Winter, R. (2015). Condensation agents determine the temperature-pressure stability of F-actin bundles. Angew. Chem. Int. Ed. Engl. 54, 11088–11092.
2. Rosin, C., Erlkamp, M., von der Ecken, J., Raunser, S., and Winter, R. (2014). Exploring the stability limits of actin and its suprastructures. Biophys J 107, 2982–2992.
3. Müller, M., Mazur, A. J., Behrmann, E., Diensthuber, R. P., Radke, M. B., Qu, Z., Littwitz, C., Raunser, S., Schoenenberger, C.-A., Manstein, D. J., et al. (2012). Functional characterization of the human α-cardiac actin mutations Y166C and M305L involved in hypertrophic cardiomyopathy. Cell. Mol. Life Sci. 69, 3457–3479.
4. von der Ecken, J., Müller, M., Lehman, W., Manstein, D. J., Penczek, P. A., and Raunser, S. (2015). Structure of the F-actin-tropomyosin complex. Nature 519, 114–117.
5. Behrmann, E., Müller, M., Penczek, P. A., Mannherz, H. G., Manstein, D. J., and Raunser, S. (2012). Structure of the rigor actin-tropomyosin-Myosin complex. Cell 150, 327–338.
6. Lehman, W., Orzechowski, M., Li, X. E., Fischer, S., and Raunser, S. (2013). Gestalt-Binding of tropomyosin on actin during thin filament activation. J. Muscle Res. Cell. Motil. 34, 155–163.
7. Behrmann, E., Tao, G., Stokes, D. L., Egelman, E. H., Raunser, S., and Penczek, P. A. (2012). Real-space processing of helical filaments in SPARX. J Struct Biol 177, 302–313.
8. Kabsch, W., Mannherz, H. G., Suck, D., Pai, E. F., and Holmes, K. C. (1990). Atomic structure of the actin:DNase I complex. Nature 347, 37–44.
9. Bowen, D., Rocheleau, T. A., Blackburn, M., Andreev, O., Golubeva, E., Bhartia, R., and ffrench-Constant, R. H. (1998). Insecticidal toxins from the bacterium Photorhabdus luminescens. Science 280, 2129–2132.
10. Waterfield, N. R., Bowen, D. J., Fetherston, J. D., Perry, R. D., and ffrench-Constant, R. H. (2001). The tc genes of Photorhabdus: a growing family. Trends Microbiol. 9, 185–191.
11. Raunser, S., Haase, W., Bostina, M., Parcej, D. N., and Kühlbrandt, W. (2005). High-yield expression, reconstitution and structure of the recombinant, fully functional glutamate transporter GLT-1 from Rattus norvegicus. J Mol Biol 351, 598–613.
12. Hobe, S., Trostmann, I., Raunser, S., and Paulsen, H. (2006). Assembly of the major light-harvesting chlorophyll-a/b complex: Thermodynamics and kinetics of neoxanthin binding. J Biol Chem 281, 25156–25166.
13. Raunser, S., Mathai, J. C., Abeyrathne, P. D., Rice, A. J., Zeidel, M. L., and Walz, T. (2009). Oligomeric structure and functional characterization of the urea transporter from Actinobacillus pleuropneumoniae. J Mol Biol 387, 619–627.
14. Bröcker, C., Kuhlee, A., Gatsogiannis, C., Kleine Balderhaar, H. J., Hönscher, C., Engelbrecht-Vandré, S., Ungermann, C., and Raunser, S. (2012). Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proceedings of the National Academy of Sciences 109, 1991–1996.
15. Behrmann, H., Lürick, A., Kuhlee, A., Balderhaar, H. K., Bröcker, C., Kümmel, D., Engelbrecht-Vandré, S., Gohlke, U., Raunser, S., Heinemann, U., et al. (2014). Structural identification of the VPS18 β-propeller reveals a critical role in the hops complex stability and function. Journal of Biological Chemistry, jbc.M114.602714.
16. The Habc domain of the SNARE Vam3 interacts with the HOPS tethering complex to facilitate vacuole fusion. (2015). The Habc domain of the SNARE Vam3 interacts with the HOPS tethering complex to facilitate vacuole fusion. 290, 5405–5413.
17. Sadian, Y., Gatsogiannis, C., Patasi, C., Hofnagel, O., Goody, R. S., Farkasovsky, M., and Raunser, S. (2013). The role of Cdc42 and Gic1 in the regulation of septin filament formation and dissociation. Elife 2, e01085.
18. Patasi, C., Godočíková, J., Michlíková, S., Nie, Y., Káčeriková, R., Kválová, K., Raunser, S., and Farkasovsky, M. (2015). The role of Bni5 in the regulation of septin higher-order structure formation. Biological Chemistry 396, 1325–1337.
19. Hernandez, J. M., Stein, A., Behrmann, E., Riedel, D., Cypionka, A., Farsi, Z., Walla, P. J., Raunser, S., and Jahn, R. (2012). Membrane Fusion Intermediates via Directional and Full Assembly of the SNARE Complex. Science.
20. Liu, Y., Petrovic, A., Rombaut, P., Mosalaganti, S., Keller, J., Raunser, S., Herzog, F., and Musacchio, A. (2016). Insights from the reconstitution of the divergent outer kinetochore of Drosophila melanogaster. Open Biol 6, 150236.
21. Petrovic, A., Mosalaganti, S., Keller, J., Mattiuzzo, M., Overlack, K., Krenn, V., De Antoni, A., Wohlgemuth, S., Cecatiello, V., Pasqualato, S., et al. (2014). Modular Assembly of RWD Domains on the Mis12 Complex Underlies Outer Kinetochore Organization. Molecular Cell 53, 591–605.
22. Friese, A., Faesen, A. C., Huis In 't Veld, P. J., Fischböck, J., Prumbaum, D., Petrovic, A., Raunser, S., Herzog, F., and Musacchio, A. (2016). Molecular requirements for the inter-subunit interaction and kinetochore recruitment of SKAP and Astrin. Nat Commun 7, 11407.
23. Basilico, F., Maffini, S., Weir, J. R., Prumbaum, D., Rojas, A. M., Zimniak, T., De Antoni, A., Jeganathan, S., Voss, B., van Gerwen, S., et al. (2014). The pseudo GTPase CENP-M drives human kinetochore assembly. Elife 3, e02978.
24. Hohn, M., Tang, G., Goodyear, G., Baldwin, P. R., Huang, Z., Penczek, P. A., Yang, C., Glaeser, R. M., Adams, P. D., and Ludtke, S. J. (2007). SPARX, a new environment for Cryo-EM image processing. J Struct Biol 157, 47–55.
25. Lymn, R. W., and Taylor, E. W. (1971). Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10, 4617–4624.